
Moving on from small molecule chemotherapies for glioblastoma
Moving on from small molecule chemotherapies for glioblastoma
It's time to go all in on other modalities

In the early 20th century, surgical intervention for glioblastoma (GB) resulted in average patient survival of 10.1 months1. Despite more than 100 years of research and billions of R&D dollars spent since2, average patient survival with the standard-of-care has increased to just 14.6 months3. These increases came after addition of radiation in the 1950s, then concomitant and adjuvant temozolomide (TMZ) in 2005.
The overwhelming focus in drug development for GB has been small molecule chemotherapies or modalities with a similar mechanism: direct interference with cancer cell division. And this hypothesis has been tested in nearly every variation imaginable. Some examples are: small molecule drugs dosed both systemically and locally; reservoirs of carmustine loaded into stiff, biodegradable wafers which were left in tumor resection cavity (Gliadel); light-activated photosensitizing agents combined with a light source added in the brain (photodynamic therapy or PDT); tumor-treating fields, where stimulating with an electric field at 100-300 kHz frequency disrupts cancer cell division4. Not one of these has been a game-changer for GB. Patients and their loved ones (and here I speak from personal experience) deserve better.
This ‘direct intervention’ hypothesis has been thoroughly tested. I find it unlikely that the next attempt at delivering such small molecule drugs – whether it be a new convection-enhanced delivery platform, slow-eluting reservoir, or nanoparticle carrier – will be successful. We have no shortage of drug delivery systems (DDS). We have a shortage of drugs worth delivering.
Our understanding of glioma biology has grown dramatically over the last two decades, and this insight should inform our therapeutic approach. I believe it is time to go all in on new modalities for GB, including protein drugs, degraders, and cell therapies.
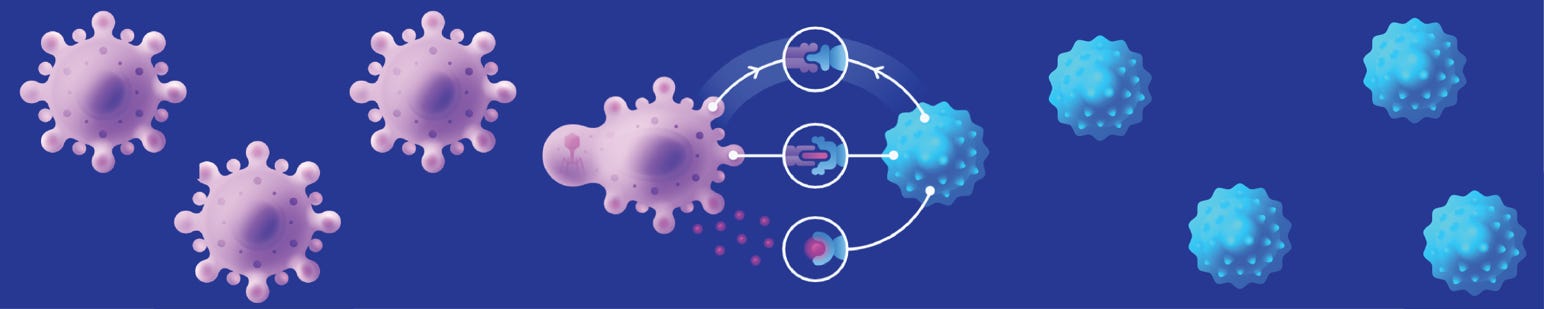
GBs polarize myeloid cells and neurons in their microenvironment to a pro-cancer phenotype, and these cells contribute to tumor growth5. My hypothesis is that reprogramming the immune and neuronal microenvironment of GBs is the key to reducing tumor burden and driving a paradigm-shift in GB patient outcomes.
Cancer immunotherapeutics, in particular checkpoint inhibitors such as Nivolumab, have dramatically improved patient clinical outcomes in aggressive stage IV melanoma. Initial attempts at using the exact same checkpoint inhibitors for GB had no clinical benefit whatsoever6. Since nearly half of the cells in a GB tumor are myeloid-derived suppressor cells (MDSCs) that sculpt an incredibly immunosuppressive microenvironment, I believe that systemically administered immunotherapies will have a much stronger effect if they are combined with local immunotherapy that targets MDSCs. There is precedent for leaving behind drugs in the resection cavity (Gliadel), and a number of companies are already working on immunotherapy reservoirs. One candidate I am particularly excited about is the inflammatory cytokine IL-12, which has the ability to reprogram MDSCs and the tumor immune microenvironment. There is preclinical data supporting IL-12 as a strong candidate for brain cancer immunotherapy (this was a large component of my PhD thesis7). Beyond protein drugs, there is also exciting clinical evidence that other modalities, like CAR T cell therapy, can have a strong effect on pediatric gliomas8 when first given intravenously then intracerebroventricularly. IL-12 will also synergize with CAR T cell immunotherapy9, including in brain tumors10. While initial attempts at brain cancer immunotherapy were unsuccessful, recent preclinical and clinical evidence warrant a deeper, venture-backed exploration of protein and cell therapies for GB.
And there’s more to a brain tumor than cancer and immune cells. Neurons also participate in glioma progression11. One mechanism by which neurons promote tumor growth is by shedding soluble neuroligin-3 (NLGN3), which has a catalyzing effect on cancer cell growth. Can antibody drugs or extracellular protein degraders (LYTACs12) slow down disease progression? There are many such modalities well poised to exploit new insights into glioma neurobiology whose full potential in the central nervous system have not yet been reached. Whether the platform companies commercializing these modalities choose to enter this space is another matter altogether.
I spent my Ph.D. thinking about these ideas in an academic setting. Having recently defended my thesis, I am now making plans for the next stage of my career. My ambition is to push new modalities for GB and neuroimmunology more broadly into the clinic. I want to build and invest in the companies that will do this.
I’ll be sharing my progress towards this ambition more regularly on Substack and Twitter; always happy to connect with anyone interested in neuroscience, neuroimmunology, and GB translational biomedicine.
McCutcheon and Preul, World Neurosurgery 149 (2021).
U.S. Department of Health and Human Services, National Institutes of Health. RePORT: Estimates of Funding for Various Research, Condition, and Disease Categories (RCDC). Retrieved January 18, 2023, from https://report.nih.gov/funding/categorical-spending#/
Stupp et al, NEJM 352 (2005).
I have met very few scientists who believe this mechanism or the reported benefits are real
Tabet et al, Front Integr Neurosci 16 (2022).
Reardon et al, JAMA Oncol 6 (2020).
Omuro et al, Neuro-Oncology 25 (2023).
Tabet et al, bioRxiv (2022).
Majzner et al, Nature (2022).
Momin et al, Sci Trans Med (2019).
Agliardi et al, Nat Comm 12 (2021).
Venkatesh et al, Cell 161 (2015).
Venkatesh et al, Nature 573 (2019).
Venkataramani et al, Nature 573 (2019).
Banik et al, Nature 584 (2020).